
Table of Contents
はじめに
前回、AlphaFoldを理解したいけど生物学系の知識がないので勉強してみた 前編:事前知識、背景理解ということで、タンパク質の構造がわかるとどうして嬉しいのかということをタンパク質とはというところから調べてまとめました。
AlphaFoldは端的にいうと 「アミノ酸配列からタンパク質の三次元構造を予測する」モデルです。 タンパク質の構造予測をするコンペである2018年のCASP13ではぶっちぎりの1位をとり、2020年のCASP14でもAlphaFold2がさらに精度を上げて話題になりました。 論文や実装が公開されているのはAlphaFold1の方なので、今回はその論文を読んできたのでまとめます。
論文のリンクはこちら Improved protein structure prediction using potentials from deep learning
概要
"タンパク質の構造予測"とはアミノ酸配列からタンパク質の三次元形状を決定することであり、これはタンパク質の機能を決定するうえで非常に重要な課題である。しかし、実験的にその構造を決定することは非常に難しい。
最近では遺伝情報を活用することで、タンパク質の構造予測にかなり進歩がみられるようになってきた。 例えば、相同配列の共分散を分析することで、どのアミノ酸残基が接触しているかを推測することができ、タンパク質の構造予測に役立つことなどがある。
これらの情報を利用することで平均力ポテンシャル を構築し、タンパク質の正確な構造予測を可能にしている。ポテンシャルはねじれ角である(φ、ψ)で微分可能なので、シンプルな勾配降下アルゴリズムで三次元構造を予測することができ、複雑なサンプリング手順は不要であることが分かった。
AlphaFoldでは相同配列が少ない場合でも高い精度を出した。 CASP13ではブラインドアセスメントにおいてフリーモデルドメインで43問のうち24個において高い精度を出した(TMスコアが0.7以上であった)。同大会で第2位の精度を出した方法ではサンプリング&コンタクト情報を用いた手法で43問のうち14問において高い精度を出していた。
AlphaFoldはタンパク質の構造予測を大幅に前進させた。 これにより、相同タンパク質の構造が実験的に決定されていない場合に、タンパク質の機能や機能不全の解明が可能になると期待される。
と論文のアブスト部分に書いてありました。 これをさらに要約すると
「相同配列の共分散を分析することで、どのアミノ酸残基が接触しているかを推測する」 これはコンタクトマップと呼ばれるものを作ることに他なりません。これはアミノ酸配列のうちi番目の残基とj番目の残基の距離がどれくらいかをというものです。これを全てのiとjの組み合わせで作成します。
最後にシンプルな勾配降下アルゴリズムでポテンシャルエネルギーが落ち着くような構造を作成するということをして予測をしているということです。
全体像を眺める
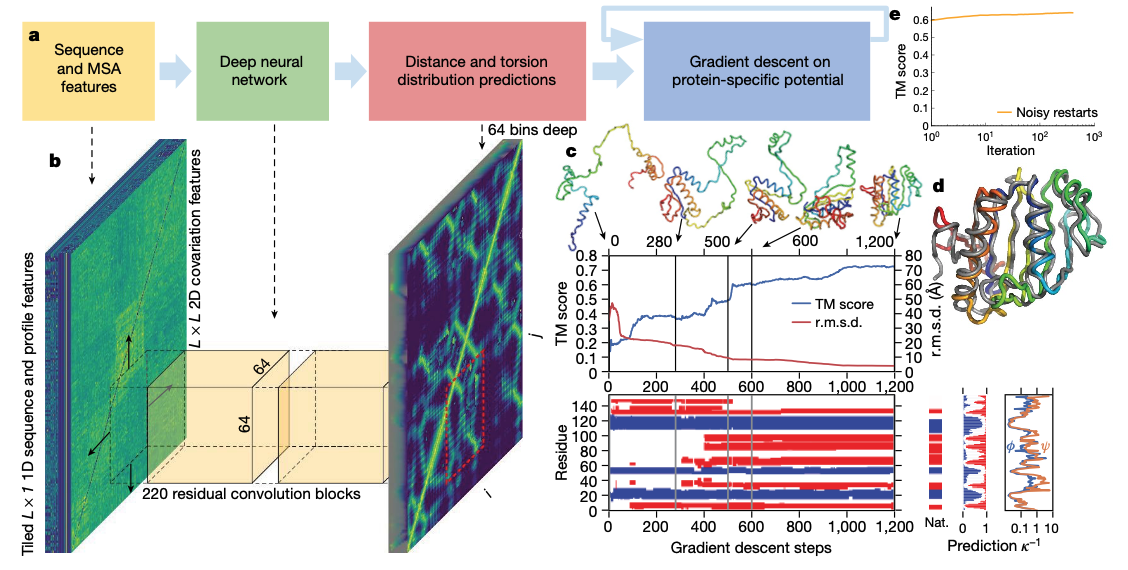
この画像は論文内のFig.2です。 これがモデルの全体像となるわけです。 細かいところは置いておいて、全体として二段階のステップを踏むようになっています。
第一段階では 「アミノ酸配列 -> 物理情報(結合距離、結合角)」
第二段階では 「物理情報(結合距離、結合角) -> 三次元構造」
を予測するように作られてます。
入力になるアミノ酸も、ただ一次元のアミノ酸配列を入力にするのではなく、類縁種のアミノ酸配列を用いてMSA(多重配列アライメント)を入力としているのが特徴ですね。そうする事により、二次元の情報として扱えるので、画像のように扱うことができるようになります。そこで、CNNを用いて特徴を抽出します。 図にもありますが220層のResidual Networkが作られていて、その出力としてコンタクトマップが出力されていますね。 図中の赤いところで「距離とねじれの分布を予測」と書いています。
そして、その物理情報を入力として第二段階に進み「勾配降下法でタンパク質のポテンシャルを最小化」を行っています。
Introduction
AlphaFoldの成績
CASPは2年に1度行われるブラインドによるタンパク質構造予測コンペで様々なグループが参加します。2018年、AlphaFoldはCASP13へ世界の97グループのうちの1グループとしてエントリーしました。 各グループは実験的に構造が決定された84個のタンパク質のそれぞれについて最大5つの構造予測を提出します。
予測する対象のタンパク質により問題のパターンがいくつかあり、テンプレートベースモデリング(TBM:類似配列を持つタンパク質が既知の構造を持ち、その相同構造を配列の違いに応じて変更する)が可能なものと、フリーモデルリング(FM:相同構造が得られないもの)が可能なものと、中間的な(FM/TBM)カテゴリーに分類します。
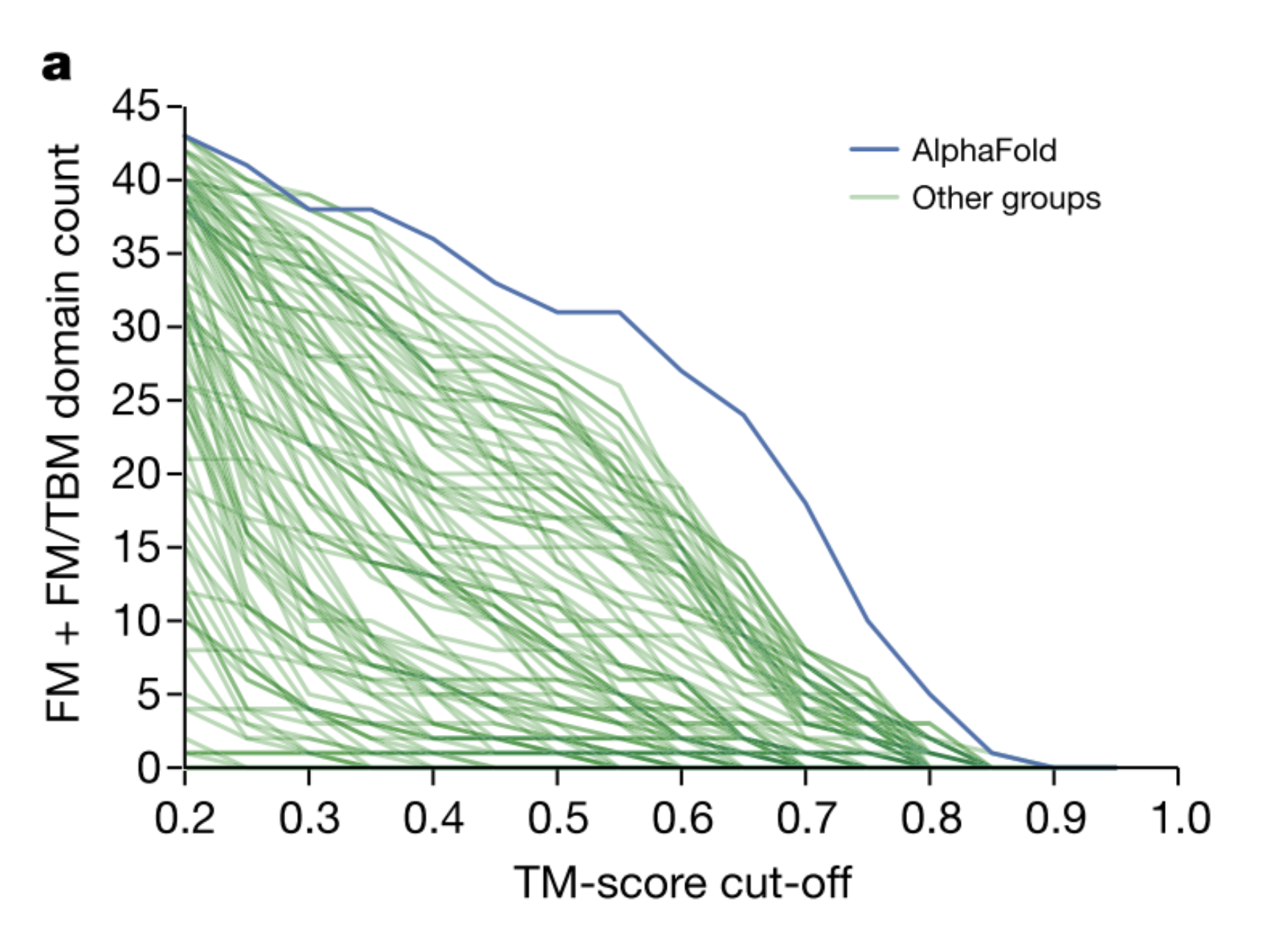
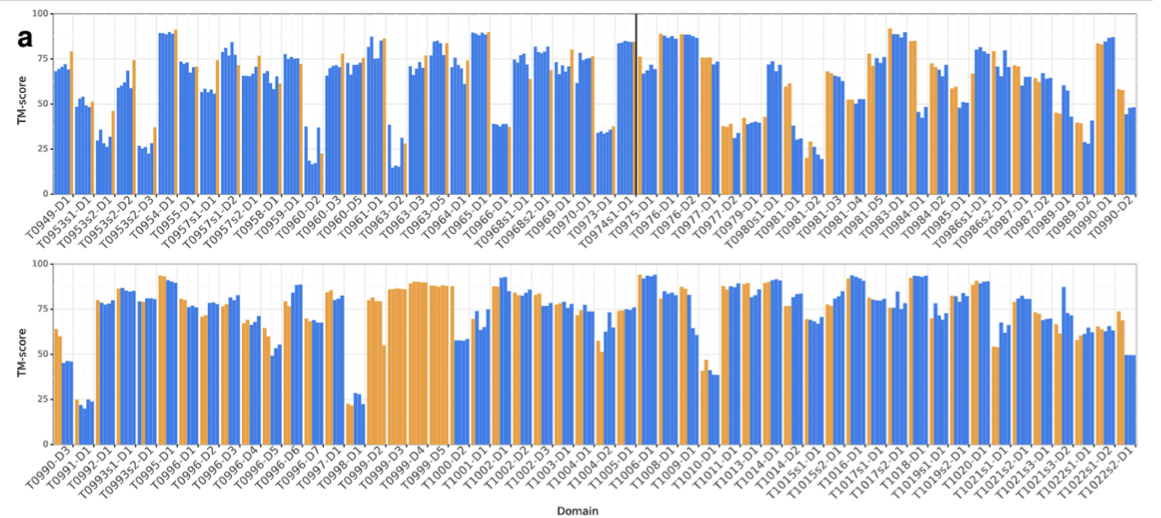
上の図.1aは、AlphaFoldが他のどのシステムよりも高い精度でより多くのFMドメインを予測していることを示しており、特に0.6〜0.7のTMスコア範囲では、より多くのFMドメインを予測していることを示していますね。FMドメインということは、ヒントがない(相同構造が得られない)状態なのにスコアが高いなんてすごい!
※ TMスコア
TMスコアは、提案された構造物の全体的なバックボーン形状がネイティブ構造物とどの程度一致しているかを測定し0から1までの範囲で表現するもの。
評価者(運営)は、98の参加グループをキャップ付きのzスコアによってランク付けしました。 FMカテゴリーにおいてAlphaFoldは、52.8の合計zスコアを達成しました。ちなみに二位は36.6でした。 ぶっちぎりの成績ですね!
zスコアは平均が0、標準偏差 (SD) が1になるように変換した得点。偏差値のイメージに近い(偏差値は真ん中が50だけど、zスコアは真ん中は0)
FMとTBM/FMのカテゴリーを合わせると、AlphaFoldは48.2点から68.3点となりました。
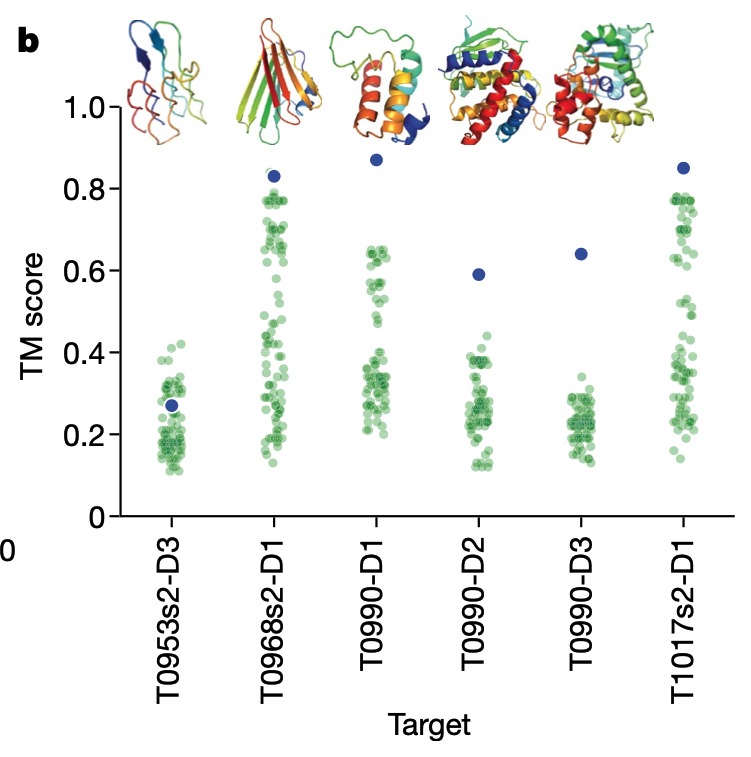
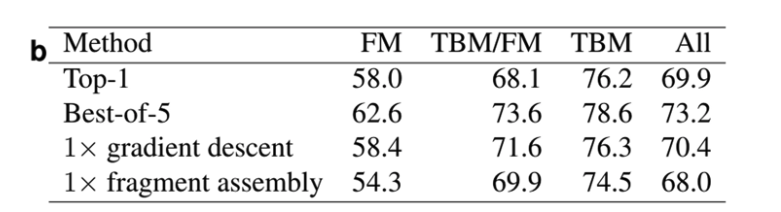
AlphaFoldではテンプレートを使用せず、FM手法のみを使用しているにもかかわらずTBMカテゴリでも高いスコアを獲得し(Fig.1b)、トップ1モデルでは4位、ベスト5モデルでは1位となりました。
この高精度を実現するのに貢献しているのは距離予測の精度によるものであり、それは対応する接触予測の精度の高さからも明らかである
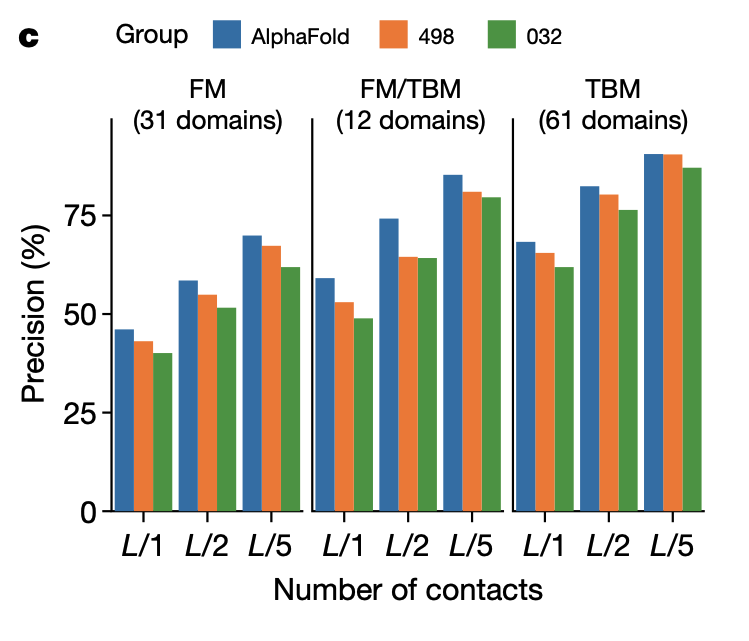
Fig.1c
従来手法
従来手法で、FMアプローチに対して最も精度が高かったのはフラグメントアセンブリ法というものだったらしいです。名前の通りフラグメント(ある一部分、破片)を操作していくことでポテンシャルエネルギーを最小化するようにサンプリングしていく手法です。 つまり、局所最適を積み上げていくことで全体最適を目指しているということですね。
精度をあげるテクニック
進化共分散データの利用することで精度が向上することがわかっています。 DNA塩基配列から得られたアミノ酸配列の大規模データセットを検索し、多重配列アラインメント(MSA)を生成することで精度向上が期待できます。 MSAの配列間の2つのアミノ酸残基の位置関係の相関関係の変化は、どの残基が接触しているかを推測するために使用することができます。というのも、類縁種のアミノ酸配列が進化の過程で一部が変化した時、空間内で近いところに位置するアミノ酸残基も呼応して変化することがあるらしく(これを共進化と呼ぶらしい)、類縁種のアミノ酸配列をアライメントした多重配列アライメントを作成して上げることで三次元空間内での残基間の距離のヒントになるとのこと。
AlphaFoldの手法
コンタクトマップの作成について
記事の前半で、第一段階では「アミノ酸配列 -> 物理情報(結合距離、結合角)」を予測するものと書きました。このパートではその部分を掘り下げていきます。
従来では「接触」について2つの残基のβ-炭素原子が互いに8Å以内にあるかないかのバイナリで表現していたそうです。しかしAlphaFoldではもっと細かい予測を行っています。Methodsのところに書かれていたのですが、コンタクトマップを作成するときは、2Å ~ 22Åを64分割したbinに分けて、そのクラスを予測する設計にしているそうです。
さらに結合角の予測に関しても工夫がされています。 普通であれば -π ~ π までの角度を等分割して予測するということが思いつきそうですが、AlphaFoldではラマチャンドランプロットを利用することで無駄な予測を省いています。
アミノ酸残基の結合角のとりうる角度(φ、ψ)について、ある組み合わせだとアミノ酸残基が三次元空間内で衝突してしまうために存在し得ない(φ、ψ)の組み合わせがあります。ラマチャンドランプロットはアミノ酸残基が存在しうる(φ、ψ)の組み合わせについて示してくれたものです。
これを用いることで、存在し得ない予測対象を省くことで精度を上げています。 (これは、ドメイン知識ありきですね。構造生物学などの専門家がチームにいるのでしょう)
少し込み入った話をします AlphaFoldの中心的なコンポーネントはPDB(Protain Data Bank)構造を学習した畳み込みニューラルネットワークで、Cβ(β炭素)の距離dijを求めるものです。タンパク質のアミノ酸配列Sと、その配列のMSA(S)から得られる特徴量に基づいて、画像認識に用いられるネットワークと似た構造の離散確率分布P(dij | S, MSA(S))を図2bに示すようにL×L距離行列の任意の64×64領域の各ij組について予測する。
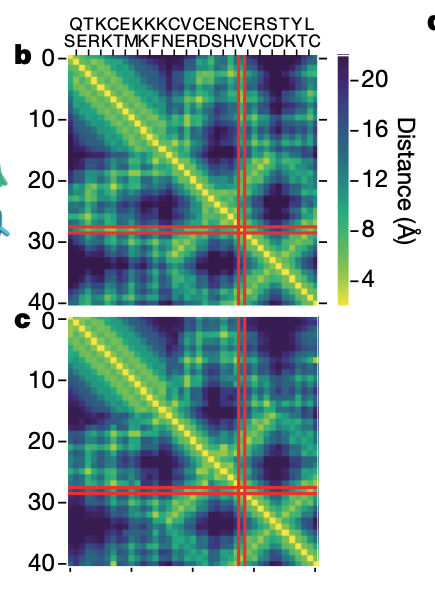
論文中の図3.bを引用
これは、CASP13で実際に使用された問題のT0955についての答え(b)と予測(c)です。 とても正確に予測できていることがわかりますね
精度を上げるためのテクニックとしてMethodsにいくつかポイントが書かれていて、
- ニューラルネットワークのデザイン
- training
- data augmentation
- feature representation
- auxiliary losses
- cropping and data curation
が挙げられていました。
特にポイントになるところとしては、アミノ酸の残基全てを入力にするのではなく64にcropしたものを入力にするところでしょう。 これは、メモリなどハードウェアの制約もあるとは思いますが、こうすることでPDBのデータを複数回利用することができある意味でのdata augmentationとしても作用しているとのこと。
論文中では
訓練でドメインが使用されるたびにcropのオフセットをランダム化することで、1つのタンパク質が何千もの異なる訓練例を生成することができるデータ増強の一形態が実現します
と書かれていました。 このdata augmentationの際のテクニックとして論文中では
エッジ効果を避けるために、生成オフセットに関しては、cropの中心付近の予測値に大きな重みを付けて平均化します。また精度をさらに向上させるために、わずかに異なるハイパーパラメータで独立して訓練された4つの別々のモデルのアンサンブルからの予測値を一緒に平均化します。
実際には微妙に違う4つのモデルのアンサンブルで予測をしていたんですね。
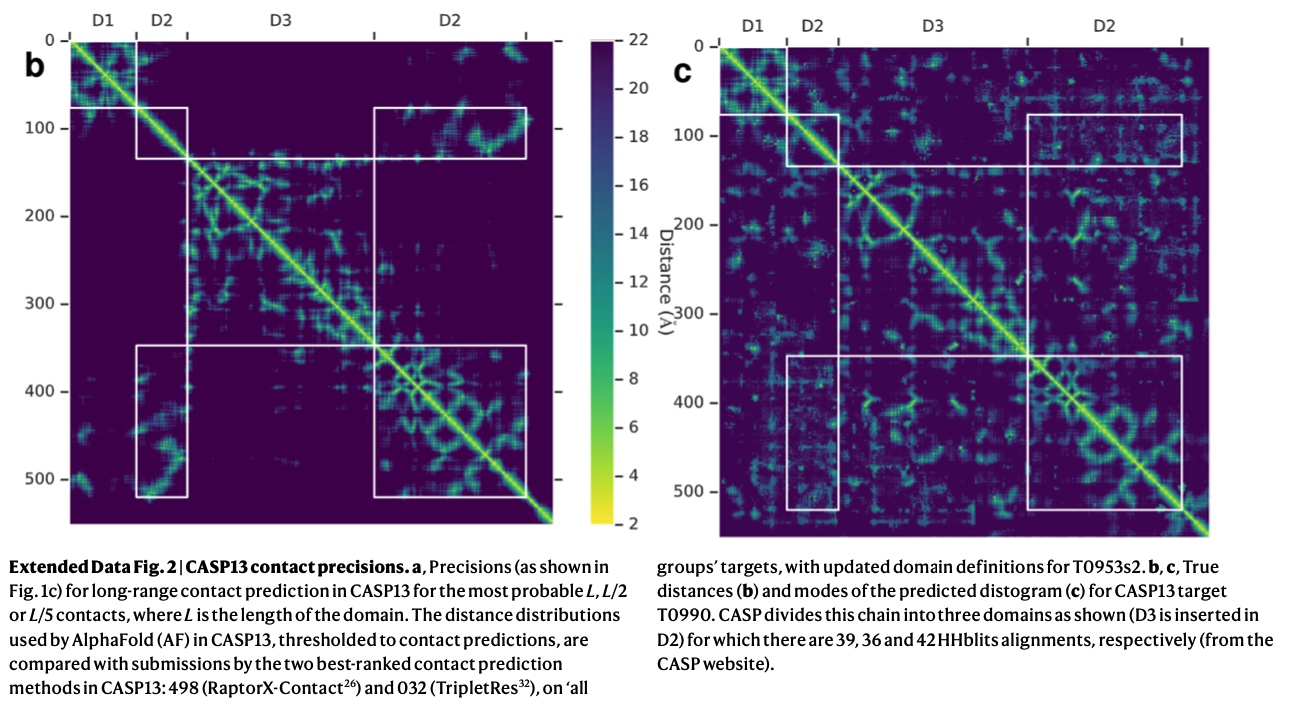
論文中のExtendedFig.2より引用
さらにトレーニング時のテクニックとしては 個々のトレーニングランについて、27個のCASP11 FMドメインを検証セットとして使用し、early stoppingを行い、交差検証をしてハイパーパラメータの調整をしたとのこと。
勾配降下法による構造の実現
第一ステップで構築されたコンタクトマップをもとに構築されたポテンシャルを最小化する構造を実現する。 理想的なタンパク質バックボーン形状の微分可能なモデルを作成し、バックボーン原子座標をねじれ角(φ,ψ)の関数として与える:x = G(φ,ψ)
ポテンシャルエネルギーの関数の各項はねじれ角に関して微分可能であるため、予測されたねじれマージンからサンプリングできるねじれの初期集合φ,ψが与えられると、L-BFGSのような勾配降下アルゴリズムを用いてポテンシャルエネルギーを最小化することができます。
最適化された構造は初期条件に依存するので、初期設定を変えて最適化を複数回繰り返します。 20個の最も低いポテンシャルの構造体のプールが維持され、一杯になったら、バックボーンのねじれに30°のノイズを加えたものから軌道の90%を初期化します(残りの10%はまだ予測されたねじれ分布からサンプリングされています)。CASP13では、チェーンごとに5,000回の最適化ランが走ったらしい。
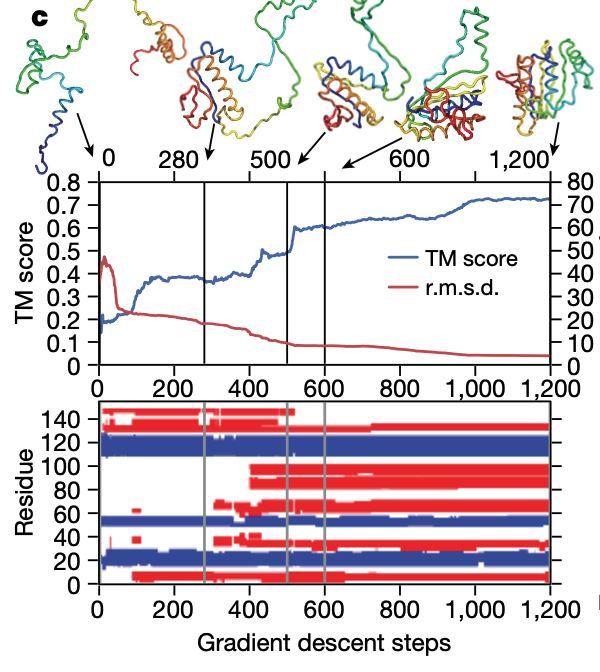
この図2.cは最適化ランとTMスコアの関係をグラフにしたもの。
また、フラグメントアセンブリ方ではなく、勾配降下法を使った方が精度が良いことをExtended Fig5. a及びbに示されている
Ext Fig 5.aの黄色のバーが勾配降下法で、青いバーがフラグメントアセンブリ法
総括
今回のAlphaFoldは生物学のドメイン知識と、機械学習の手法を巧みに織り交ぜて行われたものとしてとても感銘を受けました。 論文を読んだらみなさん気づくと思いますが、これといって機械学習的には飛び抜けて変わったことはされていません。しかし、一般的な手法を生物学のドメイン知識を使って、機械学習で扱いやすいタスクとして設計してあげるというところがすごいですね。
去年の秋ごろにCASP14も開催されて、AlphaFold2なるものがさらに精度を上げているそうです。 まだ論文や実装は公開されていませんが、transformerをベースにしたモデルにしたことでさらに精度が上がったとのこと。公開された暁にはぜひ、その論文も読んでみたいものです。
最後に
私は生物に関しては、高校の「生物基礎」で知識が止まっていて、大学受験でも「物理・化学」を選択していて、大学でも電子情報工学を専攻していたので背景知識も含めてなかなか骨の折れる内容でした。
もしかしたら、僕の解釈や理解に間違いがあるかもしれません。 お気づきになった点がありましたら、お気軽にご連絡をいただけると幸いです。
この論文を読むにあたって、生物学の背景知識を得るために様々な資料やオンラインリソースを漁りました。 その中でも、生物学の知識からスタートし、alphafoldの論文解説をされていた、白金工業fmの第31回:Alphaの系譜を持つアルゴリズム タンパク質立体構造を解く に関しては、私のような初学者の第一歩としてとても大きなガイドラインとなっていただきました。この場を借りて感謝申し上げます。
p.s.
私事ですが、白金工業fmのファンです。通勤途中やジムでトレーニングしている時にいつも聞いています。
参考文献

Ryu Ishibashi
機械学習/Vue/React/Laravelとかやってます